General MD simulations
At the most basic level, molecular dynamics use the equations of Newtonian motion to simluate systems on an atomistic level. In an all atom molecular system, starting coordinates are obtained experimentally (through crystallography, CryoEM, NMR, etc.) or built (AlphaFold, homology modeling, by hand). These coordinates are then parameterized with the forcefield of choice for the simulation. Periodic or non periodic boundaries are added and the simulation volume is solvated and populated with ions. The system is then minimized and allowed to equilibrate to the simulation temperature and pressure. From here, the simulation is allowed to run keeping various parameters constant. After sufficient time has been simulated to sample the conformational space, the trajectory can be analyzed to determine physical attributes of the system.
- These steps generally:
acquire starting model
parameterize topology file
build simulation box
minimize
equilibrate
simulate
analyze
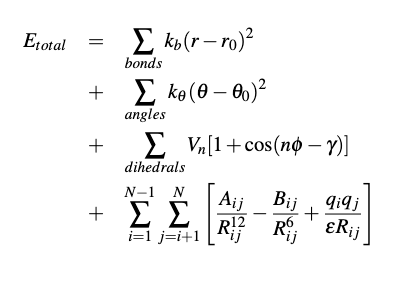
In MD simulations atoms are placed a in starting structure as points of mass and charge and given random velocities which correspond to the set temperature of the system. Atoms are then moved to their next position by calculating the force acting on each point and multiplying by the mass through the set timestep. The motion of such idealized points can be described through a set of Newtonian motion equations (right). Where the bond length and angles are modeled as springs, dihedrals through torsion calculations, and van Der Waals interactions through the Lennard-Jones potential.
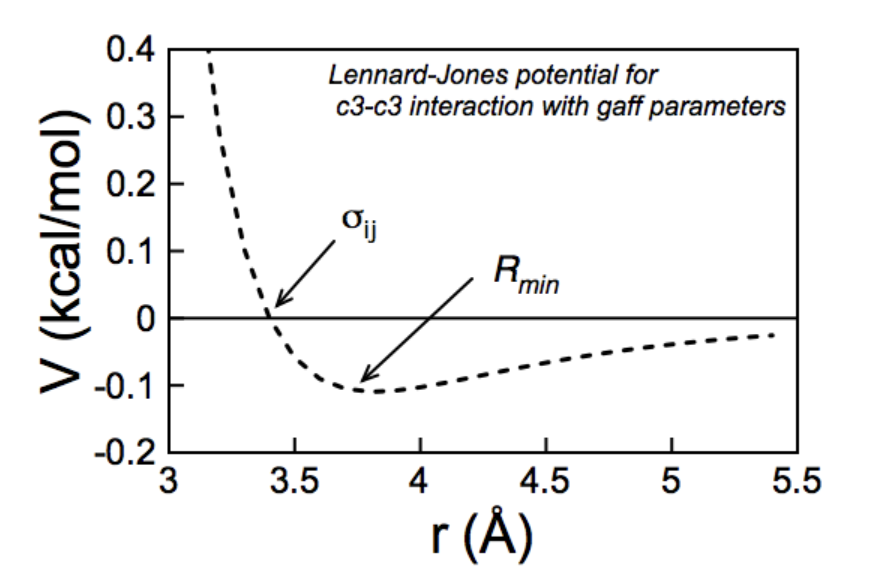
These first three terms describe the interaction of bonded atoms and thus have a finite and relatively small number of calculations. The non-bonded interactions described by the Lennard-Jones potential can, in theory describe the interaction of each atom with every other atom in the system and with the atoms in the next periodic box. As, this would lead to a prohibitively large number of calculations per timestep and as the contribution of this term decays rapidly with distance, a distance cutoff is almost always applied.